Cystická fibróza
Synonymá v širšom zmysle
Cystická fibróza, pľúca
Angličtina: mukoviscidóza, cystická fibróza
Definícia cystickej fibrózy
Cystická fibróza je dedičné ochorenie. Dedičstvo sa z lekárskeho hľadiska nazýva autozomálne recesívne. Cystická fibróza (cystická fibróza) sa nededí po pohlavných chromozómoch X a Y, ale po autozomálnom chromozóme 7.
Prečítajte si náš všeobecný článok o metabolických poruchách: Poruchy metabolizmu - čo to znamená?
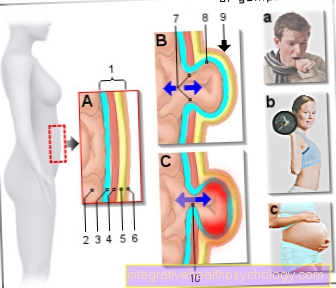
Mutácia je na tzv. Géne CFTR. Recesívne znamenalo, že na vypuknutie choroby museli byť prítomné dve poškodené kópie génu. Ak má osoba zdravé a mutované umiestnenie génov na zodpovedajúcom chromozóme 7, choroba sa nevyskytuje.
Výsledkom je patologický génový produkt. Tým sa kóduje Chloridové kanály sú rozbité. Chybné chloridové kanály vedú k tvorbe hustého hlienu vo všetkých exokrinných žľazách.
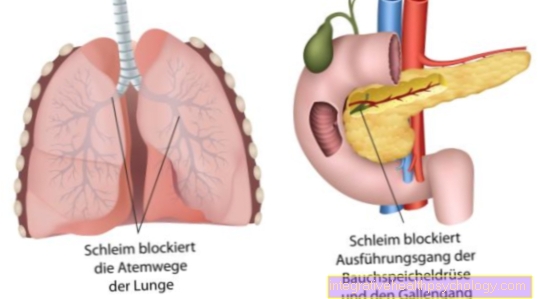
Tieto exokrinné žľazy, t. J. Žľazy, ktoré uvoľňujú svoju sekréciu von, zahŕňajú:
- pankreasu
- tenké črevo
- dýchacích ciest s pľúcami a prieduškovým systémom
- biliárny trakt a
- tiež Potné žľazy
zhrnutie
Cystická fibróza je a Dedičná choroba, Zdedil sa tak, že je rodovo nezávislý a iba s ním dva chybné gény vyskytuje. Je to najbežnejšia autozomálna recesívna dedičnosť.
Dôsledky sú tvrdé hlienové útvary všetkých exokrinných žliaz, ako sú napríklad pľúcne žľazy, pankreas a potné žľazy. Sú založené na tom narušený transport chloridu medzi vnútornou a vonkajšou časťou bunky (čítajte ďalej: Chlorid v krvi). Mutovaný gén je zapnutý Chromozóm 7 a spôsobuje široké zapojenie orgánov so zodpovedajúcimi účinkami na dýchanie, trávenie a reprodukciu.
Bohužiaľ, terapia môže zmierniť iba príznaky, ale nemôže vyliečiť. Dĺžka života u pacientov s cystickou fibrózou relatívne nízka.
Pretože ide o recesívne dedičné ochorenie, existujú ľudia, ktorí nesú zmenený gén, ale netrpia samotnou chorobou. Takéto osoby sú povolané Nosič funkcií alebo vodiče, t. j. nosiče. Títo ľudia nemajú cystickú fibrózu, pretože druhá kópia génu je neporušená a chorý nie je dosť silný na to, aby zvíťazil.
Túto chybnú kópiu génu však môže preniesť na svojho potomka. Keby už modifikovaný gén stačil na vyvolanie choroby, bolo by to takzvané dominantné dedičstvo. Takéto dedičstvo možno nájsť napríklad v Chorea huntington, Viac informácií o tejto chorobe nájdete v našej téme Chorea huntington.
Okolo 1:2500 leží Miera chorôb u novorodencov v Nemecku. dopravcu je o všetkých 25. v nemeckej populácii.
príčina

Cystická fibróza je spôsobená mutáciou génu na chromozóme 7. Tento chromozóm je autozomálny chromozóm, nie pohlavný chromozóm.
Každý má 44 autozomálnych chromozómov (dve rovnaké verzie každého) a dva pohlavné chromozómy. Táto mutácia na chromozóme 7 vedie k tvorbe defektných chloridových kanálov. Rebsorpcia (opätovná absorpcia) chloridu z žľazových sekrétov nie je možná, pretože receptor, miesto ukotvenia pre chlorid, nie je zabudované do žľazových kanálikov.
Namiesto toho je kvôli svojej nesprávnej podobe a štruktúre odložená na ťažbu. Prirodzená výmena chloridu cez určité chloridové kanály je narušená. Tieto takzvané kanály sú tvorené proteínmi. Na našej DNA je kódovaných veľké množstvo proteínov. V dôsledku genetickej poruchy chloridových kanálov dochádza k dehydratovanej a tvrdej tvorbe hlienu zo všetkých žliaz, ktoré uvoľňujú svoju sekréciu von. Hlien potom čiastočne blokuje kanáliky alebo dýchacie cesty v pľúcach.
Prečítajte si o tom tiež Mutácia chromozómov
Diagnóza cystickej fibrózy

Typickými príznakmi začínajúcimi v detstve sú priekopnícke diagnózy cystickej fibrózy.
Toto podozrenie je posilnené pozitívnou rodinnou anamnézou (choroba otca / matky alebo blízkych príbuzných). Pozitívna rodinná anamnéza znamená, že v rodine sa vyskytli alebo už boli prípady cystickej fibrózy - z hľadiska matky alebo otca.
V stolici sa dá zistiť aj nedostatok pankreatických enzýmov. Akékoľvek upchatie dýchacích ciest sa dá zistiť röntgenovým vyšetrením hrudníka.
Test na potenie, ktorý meria obsah chloridu v pote, tiež pomáha pri diagnostike cystickej fibrózy. Ak je určitá hodnota prekročená a uplatňujú sa aj ďalšie príznaky, diagnóza je relatívne pevná. Rodičia si často všimnú zvýšený obsah soli v pote dieťaťa.
Nenarodené dieťa môže byť tiež testované na toto dedičné ochorenie. Použitie punkcie plodovej vody (amniocentéza) sa odstránia fetálne bunky a skúma sa mutovaný gén.
Prečítajte si viac na tému: Röntgenové vyšetrenie dieťaťa
Liečba cystickej fibrózy
Každý, kto je postihnutý cystickou fibrózou, dostane radu v jednom Cystická fibróza - ambulancia alebo radu od Ľudský genetik (Špecialista na dedičné choroby) sa odporúča. Tieto môžu pomôcť zvýšiť kvalitu života alebo, ak chcete mať deti, vypočítať pravdepodobnosť chorého dieťaťa. Ak sú rodičia plodní a plodní.
Inak je liečba symptomatická, pretože príčinu, chybný gén, nemožno vylúčiť.
Neurčiteľná choroba
Cystická fibróza (cystická fibróza) je v súčasnosti stále nevyliečiteľnou chorobou.
V prípade cystickej fibrózy je dôležité mať primeraný príjem stolovej soli (Chlorid sodný, NaCl). Mukolýza je samozrejme zameraná. Mukolýza je rozpúšťanie hlienu, najmä v pľúcach, na uľahčenie dýchania.
Lieky a inhalácia môžu zmierniť príznaky. Ak sa funkcia pľúc výrazne zhoršuje, môže sa podať kyslík.
Intenzívnou fyzioterapiou (fyzioterapia), napríklad masáž pri poklepaní a dýchacie cvičenia, liečia sa aj pľúcne zmeny spôsobené cystickou fibrózou.
Ochorenie často končí vyžadovanou transplantáciou pľúc. Čakacie zoznamy sú však dlhé.
Súčasťou terapie je aj orálne podávanie pankreatických enzýmov a vitamínov rozpustných v tukoch. Úloha pankreasu musí byť preto podporovaná alebo skôr nahradená. Vitamíny rozpustné v tukoch sú A, D, E a K. Musia sa podávať priamo do krvi, pretože nemôžu byť absorbované z potravy kvôli nedostatku tráviacich enzýmov.
Strava by mala mať tiež vysoký obsah kalórií, pretože iba zlomok z nich možno získať z potravy.
Aby sa predišlo ďalším rizikovým faktorom komplikácií, ako je chrípka alebo zápal pľúc, dieťa by malo byť zaočkované. Odporúčajú sa nasledujúce vakcinácie:
- osýpky
- pneumokoky
- chrípka
Prečítajte si viac na tému: superinfekcie
Tieto opatrenia si, samozrejme, vyžadujú konzultáciu s lekárom, s ktorým by sa malo diskutovať o rizikách.
V súčasnosti sa v genetickom výskume kladie veľká nádej na liečbu cystickej fibrózy. Uskutočňuje sa pokus o zavedenie chýbajúcich genetických informácií do ľudského genómu. Hľadáme vektory, ktoré dokážu zvládnuť túto úlohu. Vektory môžu byť napríklad bakteriálne alebo vírusové DNA, ktoré dokážu začleniť zdravú frekvenciu do nášho genetického zloženia.
Terapeutický prístup u nenarodených pacientov sa v súčasnosti testuje. U myší sa myším embryám už podarilo zaviesť zdravý gén, ktorý obsahoval správnu génovú sekvenciu, amniocentézou (očkovaním plodovou vodou). Týmto myšiam sa teda vytvoril zdravý gén CFTR. Amniocentéza je vpich a odstránenie detských buniek z plodovej vody, čo sa vykonáva brušnou stenou matky.
V Nemecku je však táto forma vnútromaternicovej terapie (= v maternici = v maternici) zakázaná.
profylaxia
preventívne opatrenie v tomto zmysle neexistuje, pretože ide o dedičné ochorenie.
Môže sa však navštíviť centrum genetického poradenstva pre ľudí (zvyčajne sa nachádza vo univerzitných nemocniciach). Tu sa počíta, aké vysoké riziko by bolo prenesenie choroby na deti.
Táto rada je vždy užitočná, ak existuje rodinná anamnéza cystickej fibrózy.
Tiež jeden Prenatálna diagnostika stojí za to sa usilovať. Tu pred narodením (t. J. Prenatálne) a Vyšetrenie plodovej vody (amniocentéza). Z plodovej vody sa odoberú fetálne bunky (bunky od dieťaťa) a DNA sa vyšetrí na mutovaný gén.
Prognóza cystickej fibrózy
Priemerná dĺžka života u pacientov s cystickou fibrózou je, žiaľ, iba 32 - 37 rokov. V súčasnosti sa odhaduje dĺžka života novorodencov narodených s týmto ochorením okolo 45 - 50 rokov.
Prognóza do značnej miery závisí od liečby a od toho, či sa dodržiava.
Samotný pacient a jeho motivácia preto zohrávajú dôležitú úlohu.